Large scale genetic mapping studies seek to associate genetic markers, such as
SNPs
, of known location, with various quantitative and qualitative phenotypic
traits
1
. Because such fine-scale mapping requires the use of very large data sets, subsequent analysis using the
Marker-Trait Association
process often takes an inordinate amount of time. To mitigate this problem, particularly with regard to SNP data, a stripped-down version of
Marker-Trait Association
, hereafter referred to as
SNP-Trait Association
, was developed.
SNP-Trait Association
was specifically designed for very large genetic data sets. As a result, you should consider running
SNP-Trait Association
for whole
genome
scans and other large processes.
SNP-Trait Association
can output
|
•
|
|
•
|
survival curves
for a survival trait.
|
Note
: Unlike
Marker-Trait Association
,
SNP-Trait Association
does not
fully accommodate
multi-allelic markers, but
accepts
them. It creates a pseudo-bi-allelic marker using the most common
allele
as one allele and combining all other alleles into a single allele.
One
Input Data Set
is needed for this process. The
samplegmdata.sas7bdat
data set used in the following example was computer generated and consists of 1000 rows of individuals with 130 columns corresponding to data on these individuals. There are 2 categorical phenotypic
variables
(
sex
and
disease
status) and 4 quantitative phenotypic variables (
Qtrt1
,
Qtrt2
,
Qtrt3
, and
Qtrt4
).
Genotypes
for 60 different markers are presented in the two-column
allelic
format (
ma1
—
ma120
). This data set is partially shown below.
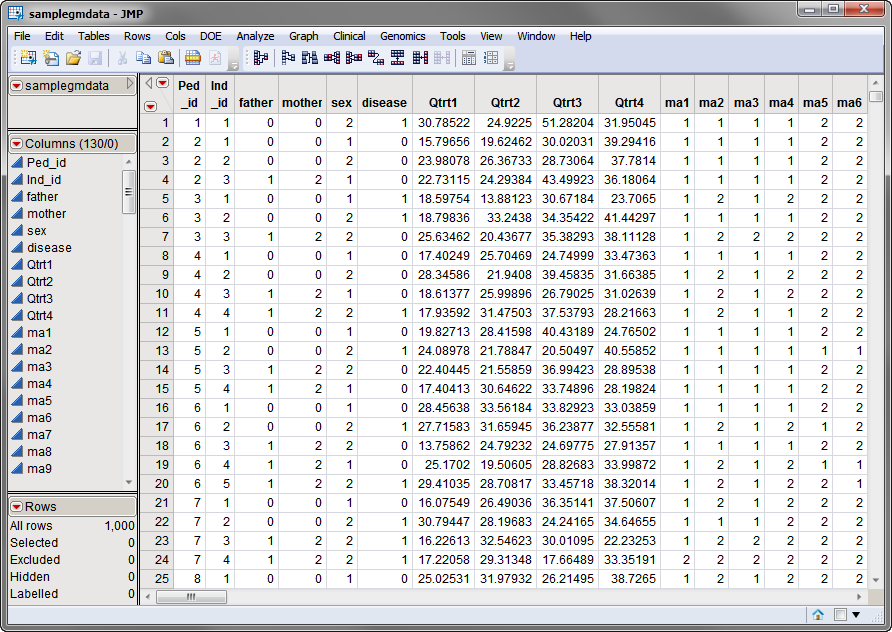
Note that this is a wide data set;
phenotypes
and markers are listed in columns, whereas individuals are listed in rows.
The second, optional, data set is the
Annotation Data Set
. This data set contains information, such as gene identity or chromosomal location, for each of the markers. The
annotation data set
used in this example, the
samplemap
data set, was computer generated and identifies markers, location and gene identities. A portion of this data set is illustrated below. This data set is a tall data set; each row corresponds to a different marker.
Note
: The top-to-bottom order of the rows in the annotation data set matches the left-to-right order of the columns in the input data set. This correspondence is required for markers to be matched appropriately.
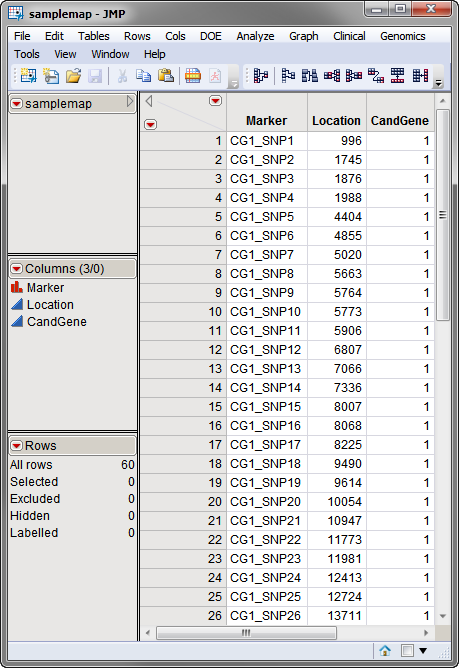
Both data sets are
described in
Data Sets Used in JMP Genomics Processes
and
are included in the
Sample Data
folder.
For detailed information about the files and data sets used or created by JMP Life Sciences software, see
Files and Data Sets
.
The output generated by this process is summarized in a Tabbed report. Refer to the
SNP-Trait Association
output documentation for detailed descriptions and guides to interpreting your results.
SNP-Trait Association
uses different SAS
PROCs
, depending on the type of traits being analyzed. You should refer to
Type of Trait
for more information.