The Build Consensus Linkage Map process imports a series of linkage maps from distinct genetic studies and uses them to estimate (via linear programming optimization) a single consensus linkage map.
Note: This map is subject to marker order constraints imposed by each input linkage map as well as optional weights for each input linkage map.
Refer to Endelman and Plomion (2014)1 for a detailed description of the method used in this process.
Two or more SAS data sets are required, one for each input linkage map. Each data set must have at least three columns; these columns must be named LinkageGroup, MarkerName, and MarkerPosition.
The LinkageGroup column lists the linkage group names or indexes (numeric or character).
The MarkerName column lists the marker names (character).
Lastly, the MarkerPosition column lists the position (in centiMorgans) of each marker on the chromosome (numerical).
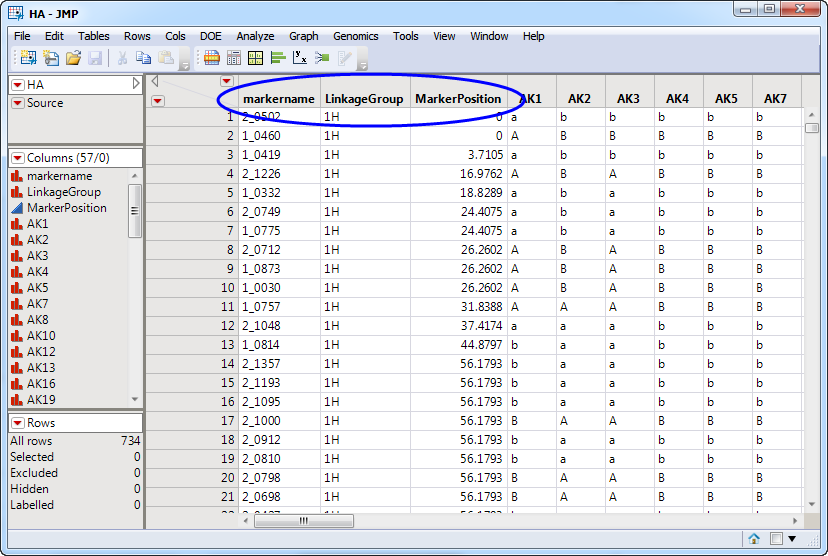
One such data set, the HA.sas7bdat data set, is shown below. Note the three required columns (circled). Additional columns list the genotypes of individual at each of the specified markers.
For detailed information about the files and data sets used or created by JMP Life Sciences software, see Files and Data Sets.
The output generated by this process is summarized in a Tabbed report. Refer to the Build Consensus Linkage Map output documentation for detailed descriptions and guides to interpreting your results.
Endelman, J.B. and Plomion, C. (2014) LPmerge: an R package for merging genetic maps by linear programming. Bioinformatics 30:1623-1624.